
 فارسی
فارسی
Definitions
Historically, the term “aplastic anemia” identifies a clinical entity where patients present with anemia and an empty marrow by bone marrow (BM) biopsy. Since 1885, when Paul Ehrlich described the first case in a pregnant woman, accumulating evidence has that this complication share several clinical, histological, molecular, and biological features, and that clinically and biologically overlap regardless of the criteria used for classification. The term “aplasia” has been replaced with BMF indicating a dramatically decrease of one or more hematopoietic lineage production leading to diminished or absent hematopoietic precursors in the BM and subsequent peripheral blood cytopenia(s).(1)
Pathophysiological Features
Direct damage to marrow
Damage to the bone marrow occurs most often iatrogenically, from chemotherapy or radiation therapy. The effects are dose dependent and transient at conventional doses. Other organ systems are affected, and spontaneous recovery is expected. Benzene also impairs hematopoiesis, and industrial exposure to benzene figured prominently in the early literature on aplastic anemia. Benzene exposure is now a negligible risk factor, accounting for only a small fraction of cases of marrow failure in most countries.1,2 However, in China, a country characterized by rapid industrialization and slower regulation, benzene remains a workplace toxin.3,4 Dosage is critical; workers with less intense or less prolonged exposure to benzene appear to have milder cytopenias, and they recover after termination of the exposure. Marrow failure is a proximate effect, not a late consequence, of benzene exposure.
Constitutional syndromes
Marrow failure results from specific loss-of-function germline mutations, usually inherited (Table 1). There is a spectrum of genetic lesions that diminish the capacity for hematopoietic stem cells to repair DNA, as in Fanconi anemia, dyskeratosis congenita and GATA2 deficiency. Marrow failure also may occur in syndromes affecting immune regulation, such as in those due to cytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) mutations8 and deficiency of adenosine deaminase 2 (DADA2).9 Constitutional syndromes are classically manifested in childhood, often with characteristic physical anomalies; typically, multiple organs are involved. The family history may disclose affected relatives. At the National Institutes of Health, among children and adults referred for protocol treatments, unexpected pathogenic mutations were very unusual in patients with severe aplastic anemia but were much more prevalent in those with moderate bone marrow failure.
Immune Aplastic Anemia
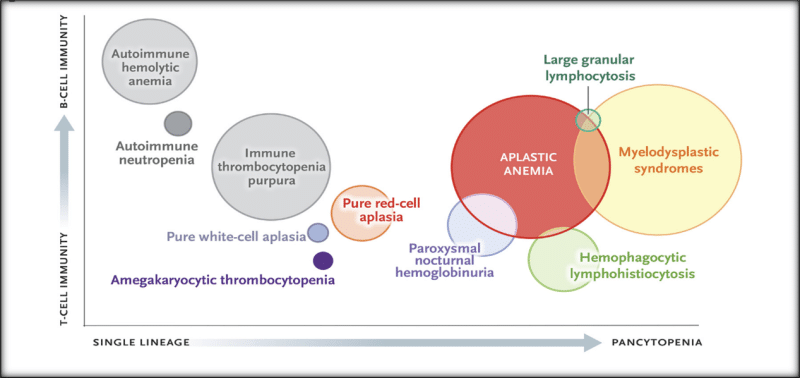
Almost all sporadic cases of aplastic anemia, especially when severe and acute, appear to be immune-mediated. The strongest, most relevant evidence for an immune mechanism is improvement in blood counts after a variety of immunosuppressive therapies and dependence of adequate counts after recovery on a maintenance calcineurin inhibitor (usually cyclosporine) Aplastic anemia is associated with immunologic diseases (particularly seronegative hepatitis, eosinophilic fasciitis, and thymoma), but most cases do not have a clear cause and have been labeled idiopathic ( Figure 1).
Diagnosis
The fatty bone marrow remains a basic diagnostic feature of aplastic anemia, but sophisticated testing can now be directed at distinguishing among diverse pathophysiological disorders and discriminating among similar, sometimes overlapping diseases in the differential diagnosis (Figure 1). Accurate diagnosis is required for appropriate therapy and effective management.
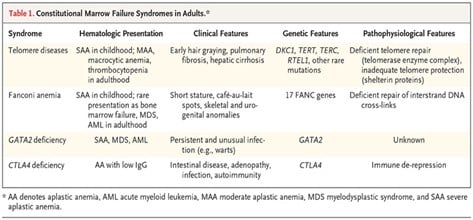
Figure (1)
Constitutional versus acquired bone marrow failure
Genomic screening complements functional testing for Fanconi anemia (indicated by chromosomal damage after clastogenic stress) and Telomeropathies (indicated by shortened Telomeres). However, comprehensive germline screening adds to the cost of the evaluation, and the results may not be available to the clinician for several weeks. Screening for the approximately 50 genes that cause constitutional marrow failure is particularly valuable in moderate and chronic pancytopenia, thrombocytopenia, and macrocytic anemia; in children and adolescents; and in patients in whom immunosuppressive therapy has failed. In patients with severe pancytopenia who do not have a family history, clinical features or evidence of organ involvement beyond the marrow, screening is not likely to be positive.
PNH and aplastic anemia syndrome
Screening for PNH is performed by means of flow cytometry, which precisely measures the proportion of GPI-anchored, protein-deficient erythrocytes and leukocytes. In classic hemolytic PNH, the PNH clone is large, above 50% and sometimes approaching representation of all circulating cells from the mutated clone. A large clone correlates with an increased risk of catastrophic venous clot and is an indication for anticomplement therapy with eculizumab, which corrects intravascular hemolysis and is effective prophylaxis against thrombosis. Clones are generally small in aplastic anemia, requiring monitoring but not treatment. Clinical PNH usually does not develop from tiny clones or in the absence of a clone at diagnosis.
Treatment
Approaches to the treatment of aplastic anemia in children and adults are shown in figure 2.
Bone marrow transplantation
Replacement of failed bone marrow is curative of the underlying disease. Historically, transplantation has been limited by its complications graft rejection and graft-versus-host disease (GVHD) — and the need for suitable donors. Expanded donor options are major recent advances.
For immune Aplastic anemia in a young patient, transplantation is always the preferred treatment. When transplantation is undertaken expeditiously after diagnosis, with the use of a graft from a histocompatible sibling donor, the results are excellent, with a long-term survival rate of more than 90% among young children and more than 80% among adolescents ,and a low rate of short- and long-term complications Although transplantation of grafts from sibling donors has become more common in older adults, the results have not improved for several decades, with a survival rate of about 50% for recipients older than 40 years of age and a relative risk of death that is almost three times as high for older adults as it is for children in registry data. Black patients also have poorer outcomes than white patients.
Also, the use of immune system suppressors and stimulation of stem cells and the use of Endogens are also other potential treatment options.
Most gratifying, treatments for patients with immune aplastic anemia have improved remarkably over the past several decades because of the development of better, transplantation and immunosuppressive regimens also Transplantation can be beneficial in all types of marrow failure.
Author and translator: Fateme Javani / Master of Hematology
Also read more:
Hematopoietic stem cell transplant failure
References